



Yoshikazu Yonei, Masayuki Yagi, and Wakako Takabe explain how glycative stress can impact health and ageing

Doshisha University Faculty of Life and Medical Sciences Graduate School of Life and Medical Sciences
email: [email protected]
Glycative stress is the condition in which various aldehydes derived from reducing sugars, lipids, and alcohol are generated in excess within the body. Among them, the most frequent cause is a glucose spike, i.e. post-prandial hyperglycemia, which, once it occurs, causes the formation of multiple kinds of aldehydes through a chain reaction (aldehyde spark) (Figure 1)1. These aldehydes — glyceraldehyde (GA), glycolaldehyde, 3-deoxyglucosone (3-DG), glyoxal (GO) and methylglyoxal (MGO) — react with the substances in the body, such as protein, and generate carbonyl modified proteins and advanced glycation end products (AGEs), enhance the secretion of inflammatory cytokine by stimulating AGEs/RAGE (receptor for AGEs) signals, and cause various disorders in cells and tissues. These reactions are known to accelerate the progression of arteriosclerosis and are closely related to the onset of cerebro-cardiovascular events.
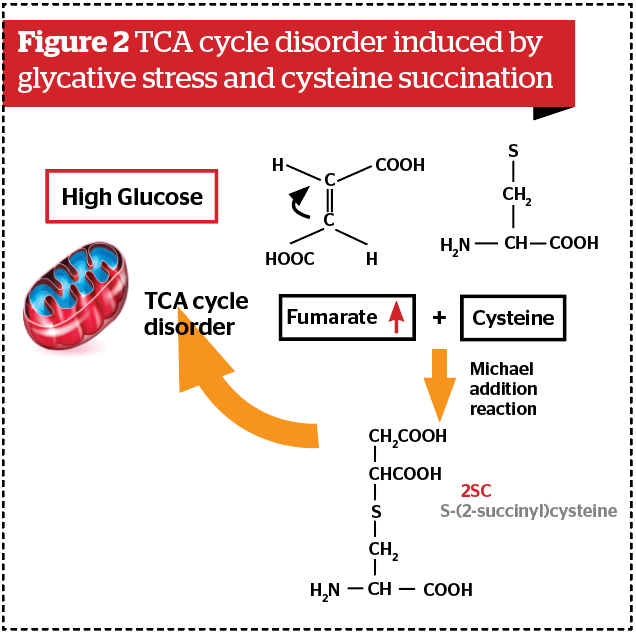
The point to be particularly emphasized is the presence of another pathway from glycative stress disorders. It has been reported that in conditions where glycative stress is substantial due to hyperglycemia or other causes, a TCA cycle disorder within mitochondria tends to occur2. If NAD (nicotinamide adenine dinucleotide) is depleted, the TCA cycle is unable to work well and fumarate is accumulated. Figure 2 shows the reaction forming by the process of cysteine succination, S-(2-succinyl)cysteine (2SC)2,3. If cysteine, which is supposed to form a physiological disulfide bond, is modified, the protein’s three-dimensional structure changes greatly. The formation of 2SC-adiponectin3, 2SC-GAPDH (glyceraldehyde-3-phosphate dehydrogenase)4 has been reported4. These two reactions are representative modifications after protein translation caused by glycative stress.
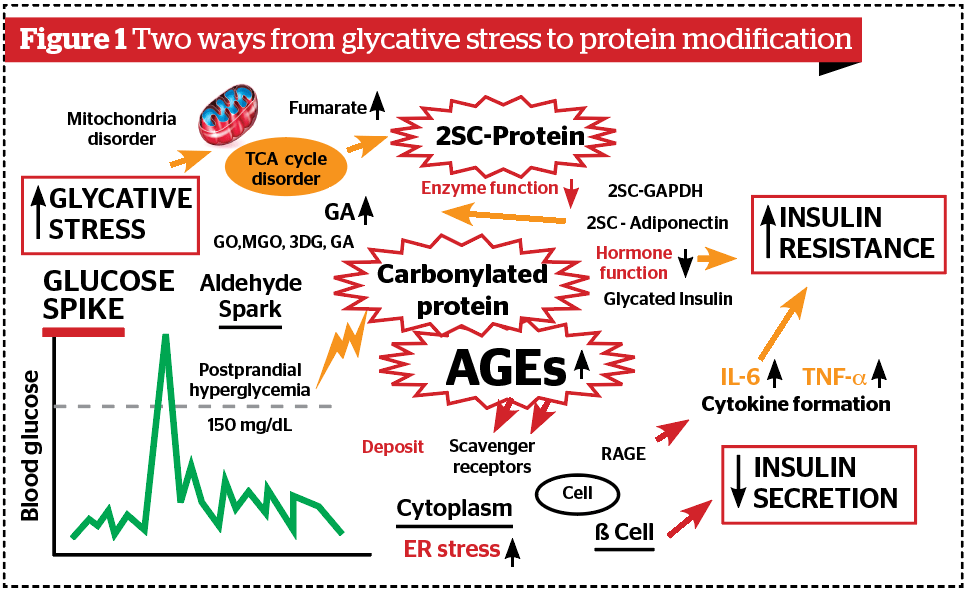
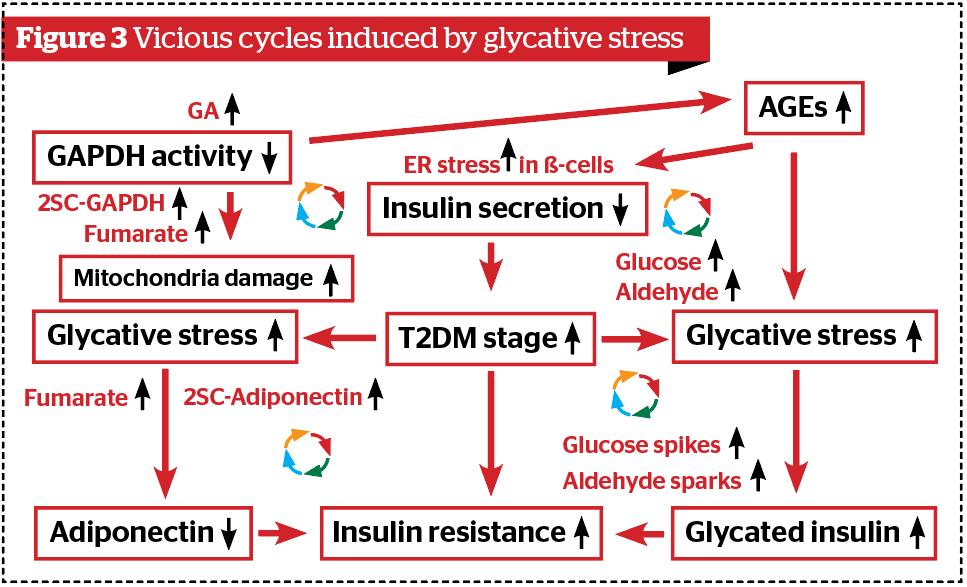
Glycative stress causes diabetic complications and various disorders, such as cataracts (glycated crystallin), dementia (glycated β-amyloid), osteoporosis (glycated type I collagen), and skin ageing (glycated collagen and elastin)1. The parentheses indicate the name of glycated proteins that plays a pathogenetic role in each disease. Furthermore, glycative stress damages the pancreas, kidney, visceral fat and skeletal muscles, and it was found that these ‘vicious’ cycles cause glycative stress to be further intensified through these disorders5. In this article, we will explain the mechanism of these cycles caused by glycative stress (Figure 3).
Insulin disorder
Type 2 diabetes mellitus (T2DM) is a representative disease in which glycative stress is high, progresses with inadequate insulin secretion, and prolonged elevated circulating glucose levels. It is confirmed through experiments that when AGEs are topically applied onto pancreatic β-cells, insulin mRNA began decreasing as well as the syntheses of proinsulin and insulin secretion5. If the additive amount of AGEs is larger, it causes apoptosis in β-cells.
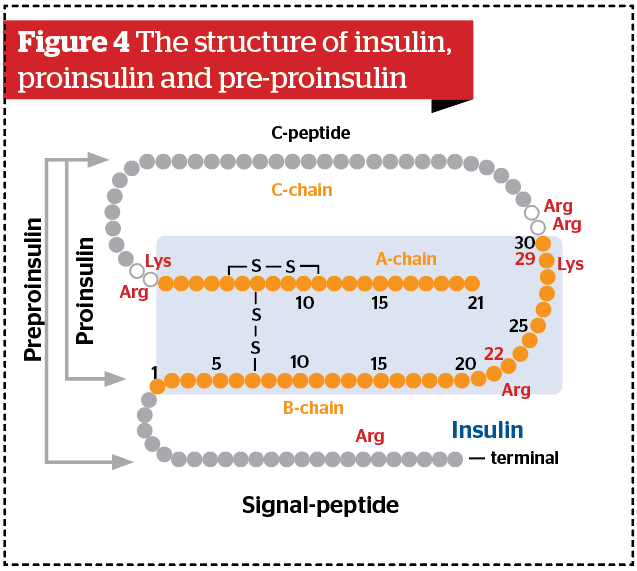
The structure of insulin, a peptide hormone, is shown in Figure 45. Initially, it is biosynthesized as pre-proinsulin in the endoplasmic reticulum (ER). The signal peptide is then cleaved off and becomes proinsulin. Proinsulin is a peptide combined with chains in the order of B chain, C chain, and A chain. After being carried to the Golgi body, proinsulin is cleaved by protease, C-chain (C peptide) is removed, and insulin is produced.
The structure of human insulin (molecular weight: 5,807) is one which A chain with 21 amino-acid residues and B chain with 30 peptide chains are combined by disulfide bonds. There is also one disulfide bond in the A chain. If 2SC is formed, A chain and B chain may not combine. In conditions with high glycative stress, the synthesis and secretion of insulin is reduced in β-cells6–8. Open-chain glucose, open-chain fructose, or aldehydes (i.e. GA, 3-DG, GO, MGO) coming from outside the cell react with pre-proinsulin and proinsulin within cells. Protein modification is then caused by carbonylation, which is then further metabolized and finally forms AGEs.
Arginine and lysine are susceptible to carbonyl modification among amino acid sequences of insulin, both of which are dibasic amino acids. This is because dibasic amino acids can retain another amino group (-NH2) even after an amino group was used for peptide bonds. We have a hypothesis that if arginine and lysine at both ends of C-peptide are modified, protease resistance increases, and as a result, C-peptide becomes unable to be disconnected and insulin synthesis decreases.
In the case of patients with T2DM, because 9% of insulin in the serum changes to glycated insulin, insulin resistance becomes elevated9. Glycated insulin has no function to uptake glucose into cells and cannot exert an insulin function9. The above facts mean that glycative stress is also involved with the onset and progression of T2DM.
Involvement of adiponectin
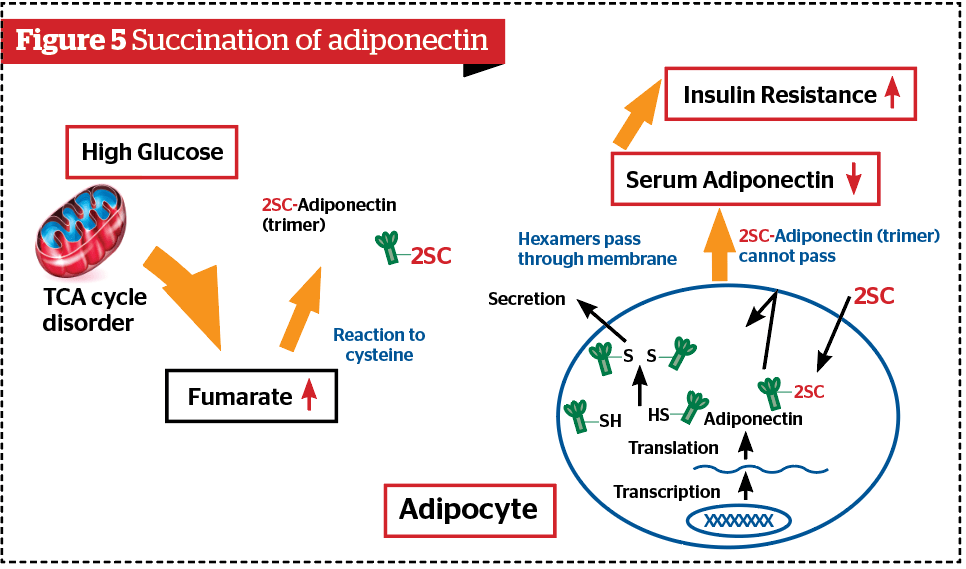
Adiponectin is one of the beneficial substances among biologically active substances (adipocytokine) secreted from the visceral adipose tissue. It exhibits actions including enhancement of fatty acid oxidation and reduction of insulin resistance and performs a preventive action against arterial sclerosis11. Adiponectin consists of three monomers and forms the structure of three chain helices (trimers) by interaction, and is stabilized by disulfide cysteine bonds. The free cysteine of the trimer formed in this way by being disulfide-bonded forms hexamers. When glycative stress increases, fumarate levels increased by the TCA cycle disorder cause 2SC modifications to trimer adiponectin and inhibit the formation of hexamer2,3. As a result, it becomes difficult for adiponectin to pass through the cell membrane, so its secretion decreases (Figure 5). This is a mechanism of another ‘vicious’ cycle included in Figure 3.
Vicious cycles through GAPDH
GAPDH is an enzyme that reduces GA by catalyzing glyceraldehyde-3-phosphate (GAP) to 1,3-bisphospho-glycerate in glycolysis and plays the most important role in the defence mechanism against glycative stress. The evidence is that cells are very rich in GAPDH, the amount of which reaches 15% to 20% of the total cytoplasmic protein. AGEs derived from GA are called toxic AGEs (TAGE)12. Cells appear to recognize that GA is toxic, so they decompose GA by forming lactate through pyruvate and excrete them.
However, if the TCA cycle is disrupted by hyperglycemia, fumarate increases and cysteine succination of GAPDH (formation of 2SC-GAPDH) occurs13,14. As 2SC-GAPDH no longer has GA metabolic activity, it cannot process GA decomposition, thus GA concentrations inside and outside of the cell increase. As a result, glycative stress is further increased. We have to realize the presence of the vicious cycle through 2SC-GAPDH (Figure 3).

Conclusion
Glycative stress causes a 2-way modification of proteins (i.e. AGE-protein and 2SC-protein) and increases the risks of T2DM and related disorders. We should learn more about the existence of these vicious cycles and implement countermeasures against glycative stress at an early stage in order to prevent the onset and progression of T2DM by ceasing these cycles and maintain our health.
Declaration of interest None
Figures 1–4 © Dr Yonei
References
- Yonei Y, Yagi M, Takabe W. Glycative stress and sleep quality. Prime: International Journal of Aesthetic & Anti-Ageing Medicine. 2018; 8(6): 19-23.
- Nagai R, Brock JW, Blatnik M, et al. Succination of protein thiols during adipocyte maturation: A biomarker of mitochondrial stress. J Biol Chem. 2007; 282(47): 34219-34228.
- Frizzell N, Rajesh M, Jepson MJ, et al. Succination of thiol groups in adipose tissue proteins in diabetes: Succination inhibits polymerization and secretion of adiponectin. J Biol Chem. 2009; 284: 25772-25781.
- Blatnik M, Frizzell N, Thorpe SR, et al. Inactivation of glyceraldehyde-3-phosphate dehydrogenase by fumarate in diabetes: formation of S-(2-succinyl)cysteine, a novel chemical modification of protein and possible biomarker of mitochondrial stress. Diabetes. 2008; 57(1): 41-49. doi: 10.2337/db07-0838
- Yonei Y, Yagi M, Takabe W. Stop the “Vicious Cycle” induced by glycative stress. Glycative Stress Res. 2020; 7(1): 13-21. doi: 10.24659/gsr.7.1_13.
- Zhao Z, Zhao C, Zhang XH, et al. Advanced glycation end products inhibit glucose-stimulated insulin secretion through nitric oxide-dependent inhibition of cytochrome c oxidase and adenosine triphosphate synthesis. Endocrinology. 2009; 150(6): 2569-2576. doi: 10.1210/en.2008-1342.
- Puddu A, Storace D, Odetti P, et al. Advanced glycation end-products affect transcription factors regulating insulin gene expression. Biochem Biophys Res Commun. 2010; 395(1): 122-125. doi: 10.1016/j.bbrc.2010.03.152.
- Shu T, Zhu Y, Wang H, et al. AGEs decrease insulin synthesis in pancreatic β-cell by repressing Pdx-1 protein expression at the post-translational level. PLoS One. 2011; 6(4): e18782. doi: 10.1371/journal.pone.0018782.
- Hunter SJ, Boyd AC, O’Harte FP, et al. Demonstration of glycated insulin in human diabetic plasma and decreased biological activity assessed by euglycemic-hyperinsulinemic clamp technique in humans. Diabetes. 2003; 52(2): 492-498.
- Boyd AC, Abdel-Wahab YH, McKillop AM, et al. Impaired ability of glycated insulin to regulate plasma glucose and stimulate glucose transport and metabolism in mouse abdominal muscle. Biochim Biophys Acta. 2000; 1523(1): 128-134.
- Kadowaki T, Yamauchi T, Kubota N. The physiological and pathophysiological role of adiponectin and adiponectin receptors in the peripheral tissues and CNS. FEBS Lett. 2008; 582(1): 74-80.
- Takeuchi M, Takino JI, Sakasai-Sakai A, et al. Toxic AGE (TAGE) theory for the pathophysiology of the onset/progression of NAFLD and ALD. Nutrients. 2017; 9(6). doi: 10.3390/nu9060634.
- Blatnik M, Thorpe SR, Baynes JW. Succination of proteins by fumarate: mechanism of inactivation of glyceraldehyde-3-phosphate dehydrogenase in diabetes. Ann N Y Acad Sci. 2008; 1126: 272-275. doi: 10.1196/annals.1433.047.
- Adam J, Ramracheya R, Chibalina MV, et al. Fumarate hydratase deletion in pancreatic β cells leads to progressive diabetes. Cell Rep. 2017; 20(13): 3135-3148. doi: 10.1016/j.celrep.2017.08.093




{kind=link}