


Nadiia Kryzhanovska, MD, PhD, reviews the literature surrounding NAD+ as a potential therapy to tackle the onset of ageing

email: nadiiakryzhanovska@ gmail.com
In 2015, KAEBERLEIN ET AL. described the primary preventive methods against age-associated diseases1. They included dietary restriction, exercise, mechanistic targets for rapamycin (mTOR) inhibitors, metformin and acarbose, NAD precursors and sirtuin activators, modifiers of senescence and telomere dysfunction, hormonal and circulating factors, and mitochondria-targeted therapeutics1. In this article, we will look at NAD precursors and treatments.
Having been discovered by Harden and Young in 19062, nicotinamide adenine dinucleotide (NAD+) brought the Nobel prize to Dr Hans von Euler-Chelpin in 19293. NAD+ was discovered as a ‘cozymase’ necessary for fermentation. Later we got to know it is a cofactor for other enzymes involved in cellular energy metabolism, as well as for the adaptive responses of cells to bioenergetic and oxidative stress, such as glycolysis, fatty acid b-oxidation, and the Krebs cycle. At the same time, the reduced form of NAD+ (NADH) is a primary hydride donor in the production of adenosine triphosphate (ATP) via anaerobic glycolysis and mitochondrial oxidative phosphorylation4.
Nowadays, the importance of NAD+ has expanded to that of a top regulator of numerous cell signalling pathways, playing a major role in ageing and age-related diseases5,6.
The majority of findings from recent studies links compromised NAD+ to ageing-associated traits (hallmarks). First, there may be impaired autophagy and mitophagy in human cells and animal models, resulting in mounting mistakes in lysosome-targeting and recycling mechanisms. Second, this age-associated deterioration results in the accumulation of damaged molecules and mitochondria; therefore, this intracellular and extracellular waste leads to cell dysfunction or death7,8.
AD+ is consumed in many catabolic pathways — in the cytosol, NAD+ is reduced to NADH by lactate dehydrogenase during anaerobic glycolysis9. In mitochondria, the three Krebs cycle enzymes, isocitrate dehydrogenase, a-ketoglutarate dehydrogenase, and malate dehydrogenase, reduce NAD+ to NADH. And NADH serves as the primary means of reducing complex I (NADH dehydrogenase) to fuel oxidative phosphorylation, generating NAD+, and ultimately reducing oxygen to H2O and producing adenosine triphosphate10.
Sources of cellular NAD+ in mammals
The greatest trait of NAD+ is its ability to be metabolised in the
gut and synthesised again within cells11.
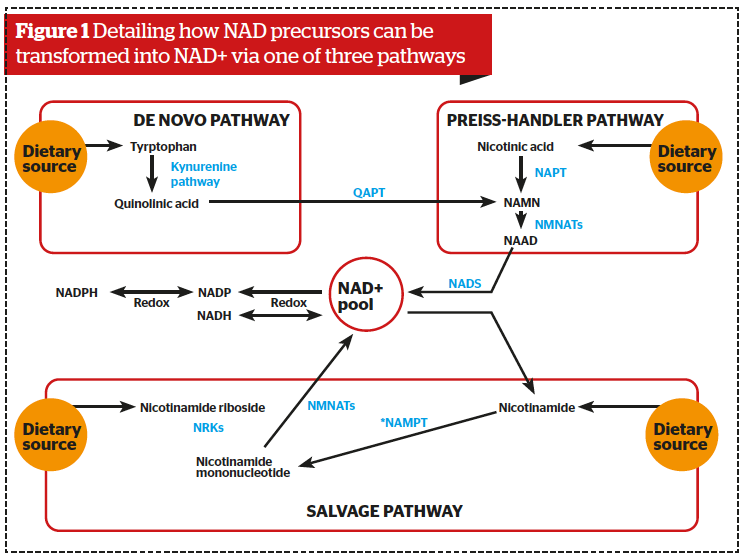
NAD+ is synthesised from the following precursors: tryptophan (Trp), nicotinic acid (NA), nicotinamide riboside (NR), nicotinamide mononucleotide (NMN), and nicotinamide (NAM)12.
Food sources of NAD precursors include fish, poultry, beef, mushrooms, brown rice, peanuts, green peas, and avocados. These precursors can be transformed into NAD+ via one of three pathways within cells, depending on the bioavailability of the precursors:
- From tryptophan (Trp) by the de novo biosynthesis pathway or kynurenine pathway
- From nicotinic acid (NA) in the Preiss–Handler pathway
- From niacinamide (NAM), nicotinamide riboside (NR), and nicotinamide mononucleotide (NMN) in the salvage pathway (Figure 1).
As mentioned above, NAD+ serves as a substrate for numerous key enzymes involved in the ageing process.
First of all, it is the group of sirtuins (SIRTs) that promote mitochondrial homeostasis, neuronal recovery, stem cell rejuvenation, and prevent certain aspects of the ageing process9,13.
The second, ADP ribosylation by poly (ADP-ribose) polymerase (PARP), is an important protein post-translational modification that affects DNA repair, epigenetic regulation, neurodegeneration, and ageing15. The PARPs are prominent members of the ADP-ribosyltransferase family, which transfer the first ADP-ribose unit from NAD+ to target proteins, followed by the sequential addition of ADP-ribose units to the preceding ones to form poly (ADP-ribose) polymers (PARs)14.
The third, CD38, is a transmembrane protein localised to the plasma membrane and membranes of intracellular organelles, including the mitochondria, nucleus, and endoplasmic reticulum15,16. CD38 is expressed in immune cells, the liver, testis, kidneys, and the brain. It plays an important role in several physiological processes, such as nuclear Ca2+ homeostasis, immunity, inflammation, as well as glucose and lipid homeostasis16. NAD+ consuming enzymes regulate a spectrum of cellular activities, including mitochondrial maintenance, DNA repair, and stem cell rejuvenation, processes that are critical for cellular health7, 17.
There is strong evidence of decreased NAD+ in aged human tissue, and it declines with age in multiple types of tissues, which include the liver, skeletal muscle, adipose tissue, heart, brain, kidney, pancreas, lungs, spleen, and skin4,18. By the time an individual is in their 50s, levels of NAD+ have fallen to half of their youthful levels, with resulting loss of sirtuins and PARP activity. Indeed, overexpression of the NAD+ biosynthetic genes NPT1 and PNC1 in yeast extends lifespan by over 50%19.
Looking again at CD38, a membrane-bound hydrolase that has been implicated in immune responses and energy metabolism. Mice lacking CD38 or treated with the CD38 inhibitor apigenin have elevated levels of NAD+ and some protection against the harmful effects of a high-fat diet20.
In addition, levels of CD38 increased in bodily tissue during the ageing process20. CD38 has been blamed for the degradation of not only NAD+ in vivo but also nicotinamide mononucleotide (NMN). When CD38 inhibited mice were given injections of NAD+, NMN or NR, circulating levels of NAD metabolites remained stable after 150 minutes, long after they began to fall in the control-type animals. So, CD38 inhibition might be the next level in NAD+ treatment16.
NAD precursors for NAD+ deficiency
NMN seems to be an efficient NAD+ precursor. In mammals, NMN is synthesised from nicotinamide, a water-soluble form of vitamin B3 and 5-phosphoribosyl 1-pyrophosphate. In addition, it can also be synthesised from nicotinamide riboside (NR), via NR kinase-mediated phosphorylation reactions.
Administration of NMN in mammals enhances the biosynthesis of NAD+ in various peripheral tissues, including the liver, pancreas, adipose tissue, heart, skeletal muscle, kidneys, eyes, and blood vessels6,21.
Furthermore, NMN was revealed to increase NAD+ in the hypothalamus and hippocampus following an intraperitoneal injection; therefore, it has properties for passing through the blood-brain barrier22. The long-term oral administration of NMN (up to 300 mg/kg) had no obvious harmful or toxic effects in normal wild mice12. And recent data showed its safety in humans23.
NMN protects from age-associated functional decline by improving energy metabolism, insulin sensitivity, lipid metabolism, mitochondrial function, eye function, bone density, and immune function18. Furthermore, it suppressed age-related changes in gene expression and adipose tissue inflammation12.
NR is another natural NAD precursor. It can be converted directly into NMN via the activity of kinases, thereby skipping the requirement of the NAMPT in the salvage pathway, and therefore serves to provide an additional pathway for NAD elevation. As NMN, NR also exhibited beneficial effects in protection against age-related diseases. It has been shown to promote longevity as well as improve healthspan in multiple laboratory animal models24.
In age-related diseases, particularly obesity, diabetes, and cardiovascular conditions, NR was able to decrease the levels of fat tissue and improve glucose tolerance25. In in vivo models of Alzheimer disease and Parkinson’s disease, NR was linked to improving memory, learning, motor function, and mitochondrial function as well as protected against neuronal cell loss26,27.
Considering DNA damage was reduced following chronic administration of NR due to its action on NAD consuming PARPs, we can begin to conclude it may be a possible treatment of neuroinflammation and mitochondrial dysfunction27. Mitophagy was suggested to be a possible mechanism promoted by NR, which can be the basis of improved mitochondrial quality29.
NAM is one more precursor for NAD and a key molecule involved in energy metabolism. In one study, it was able to restore glucagon storage, as well as ameliorate non-alcoholic fatty liver disease, oxidative stress and inflammation30. In addition, NAM has demonstrated a mitoprotective effect, especially in relation to age-associated and high-fat diet-induced DNA damage30. Its administration is tightly linked with age-dependent ophthalmological pathology.
De novo synthesis of NAD+ depends upon the quantity of Trp present and is attributed to the kynurenine pathway (Figure 1). Of note, NAD+ levels have been described as very volatile; therefore, the de novo synthesis of NAD+ plays an important role in maintaining adequate NAD+ levels. Quinolinic acid, a product of Trp degradation, was found to be essential for de novo synthesis of NAD+ and may serve as a precursor of NAD+ in the tryptophan/kynurenine pathway. However, quinolinic acid is neurotoxic, and this nullifies the positive effect of sufficiently high doses of Trp on synthesis31.
Non-precursors of NAD elevation
As mentioned above — inhibition of CD38 increases the levels of NAD+, especially in times of inflammation.
Treatment of cells with apigenin or quercetin inhibits CD38 and promotes an increase in intracellular NAD+ levels. An increased NAD+ level decreases protein acetylation through sirtuin activation. This was witnessed in one such study when treatment of obese mice with apigenin resulted in CD38 inhibition, higher NAD+ levels in the liver,
and a decrease in protein acetylation17.
One more interesting inhibition effect is regarding PARPs — the NAD+ consuming enzymes. PARPs cut NAD+ into NAM and ADP-ribose (ADPR), generating a chain. PARP1 is the most abundant of the PARPs, a major consumer of NAD+ in response to DNA injury32. PARPs and sirtuins compete for NAD+ as a substrate. PARP activity increases with the inevitable process of ageing, possibly due to the accumulation of DNA damage. Consequently, the NAD+ pool is depleted, resulting in reduced sirtuin activity32.
Conversely, pharmacological and genetic inhibition of PARPs revealed increased NAD+ content and enhanced the activity of sirtuins33. Furthermore, elevated sirtuin activity was associated with increased mitochondrial content and oxidative metabolism as well as protection against metabolic dysfunction, DNA damage, and neurodegeneration9.
NNMT gene inhibition is one more way to increase NAD+. Nicotinamide N-methyltransferase (NNMT) catalyses the methylation (switching-off) of NAM converting enzyme genes. NNMT has been shown to be involved in various disease conditions, such as metabolic disorders, neurodegenerative diseases, and cancer34. In conditions such as obesity and diabetes, NNMT levels have been reported to be upregulated significantly and genetic inhibition, as well as molecular blocking of NNMT, was shown to be beneficial in obesity protection in rodents35.

The simplest way and the hardest part
Exercise and diet can affect NAD+ concentration Exercise and diet can affect NAD+ concentration Exercise and diet can affect NAD+ concentration increase muscle NAD+ levels in a high-fat diet (HFD)-induced obesity mouse model36,37.
A number of studies in both mice and humans have shown that exercise can change NAD+ levels in a biphasic manner, calling for an increase in moderate-intensity exercise and a reduction in strenuous exercise. Highlighting the level of exercise intensity matters38.
In addition, while a high-fat diet decreases the quantity of muscle NAD+ in obese mice, exercise
and caloric restriction can increase NAD+ levels in the muscles and liver36,37. Caloric restriction induces an increase in the NAD(+)/NADH ratio in cells and results in the activation of SIRT1, an NAD(+) dependent protein deacetylase that is thought to be a metabolic master switch linked to the modulation of lifespans.
Therefore, intracellular NAD+ is not only regulated by many cellular activities, including oxidative phosphorylation, mitochondrial metabolism, transcription and signalling but can also be significantly influenced by diet and exercise.
Clinical future
In recent studies, NAD+ precursors have demonstrated their ability to elevate the blood concentration of NAD+ in healthy individuals in a dose-dependent manner and without any toxic effects39. In a 52-year-old male, NR in the dosage of 1000 mg over a period of seven days resulted in a 2.7 fold increase in blood concentrations of NAD+ and led to a 45.5-fold increase in nicotinic acid adenine dinucleotide phosphate40. Positive results of chronic administration of NAD+ precursors in animal models have led to studies in humans23,41,42 with no serious adverse effects. Thereby implicating the chronic administration of NR as a safe and effective way to increase NAD+ levels. However, safety is paramount, and as a potent cellular activator, NAD+ levels should be elevated with accuracy43.
Conclusion
Taking into consideration the effect of the NAD+ sirtuin pathway on mitochondrial and metabolic homeostasis, new supplementation strategies (using NAD, NR, NAM or NMN) look promising as a potential way to increase endogenous NAD+ availability in the treatment of age-related diseases and ageing. Carefully designed human clinical studies are required to examine these compounds further before we can propose them as valuable nutraceuticals to counteract■
References
- Kaeberlein, M., Rabinovitch, P. S. & Martin, G. M. Healthy aging: The ultimate preventative medicine. Science 350, 1191–1193 (2015).
- Harden, A. and Young, W.J. The alcoholic ferment of yeast-juice. Part II. The coferment of yeast-juice. Proc. R. Soc. Lond. 1906. B 369–375
- Euler-Chelpin, H.V. Fermentation of sugars and fermentative enzymes. In Nobel Lectures, Chemistry 1922–1941, 1966. pp. 144–155, Elsevier
- K.L.Bogan,C.Brenner. Biochemistry: Niacin/NAD(P).Encyclopedia of Biological Chemistry. 2nd Edition. 2013
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science. 2015. 350, 1208–1213
- Bonkowski, M.S. and Sinclair, D.A. Slowing ageing by design: the rise of NAD+ and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016. 17, 679–690
- Fang EF, Lautrup S, Hou Y, Demarest TG et al. NAD+ in Aging: Molecular Mechanisms and Translational Implications. Trends Mol Med. 2017 Oct;23(10):899-916
- Fang, E.F. et al. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell. 2014. 157, 882– 896
- Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014 Aug;24(8):464-71.
- Belenky, P. et al. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+. Cell 2007. 129, 473–484
- Rajman L, Chwalek K, Sinclair DA. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018 Mar 6;27(3):529-547.
- Fang EF. Mitophagy and NAD(+) inhibit Alzheimer disease. Autophagy. 2019 Jun;15(6):1112-1114
- Ramsey, K.M. et al. Age-associated loss of Sirt1-medi- ated enhancement of glucose-stimulated insulin secretion in beta cell-specific Sirt1- overexpressing (BESTO) mice. Aging Cell 2008 (7), 78–88
- Rouleau, M. et al. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010 (10), 293–301
- Aksoy, P. et al. Regulation of intracellular levels of NAD: a novel role for CD38. Biochem. Biophys. Res. Commun. 2006. 345, 1386–1392
- Escande, C. et al. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Dia- betes 2013. 62, 1084–1093
- Canto, C. et al. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 2015. 22, 31–53
- Chalkiadaki, A. and Guarente, L. The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer 2015, 608–624
- Anderson, Rozalyn M et al. “Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae.” Nature, 2003. vol. 423,6936: 181-5.
- Juliana Camacho-Pereira, Mariana G. Tarragó,Claudia C.S. Chini et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab 2016. 23, P1127-1139
- Uddin, G.M. et al. Head to head comparison of short- term treatment with the NAD(+) precursor nicotinamide mono- nucleotide (NMN) and 6 weeks of exercise in obese female mice. Front. Pharmacol. 2016. 7, 258
- Weiqi Hong et al. Nicotinamide Mononucleotide: A Promising Molecule for Therapy of Diverse Diseases by Targeting NAD+ Metabolism Front. Cell Dev. Biol., 28 April 2020
- Junichiro Irie 1, Emi Inagaki 2 3 4, Masataka Fujita. Effect of oral administration of nicotinamide mononucleotide on clinical parameters and nicotinamide metabolite levels in healthy Japanese men. Endocr J. 2020 Feb 28;67(2):153-160.
- Barbosa, M.T. et al. The enzyme CD38 (a NAD glycohydrolase) is necessary for the development of diet-induced obesity. FASEB J. 2007. 21, 3629–3639
- Camacho-Pereira, J. et al. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3- dependent mechanism. Cell Metab. 2016. 23, 1127–1139
- Long, A. et al. CD38 knockout mice show significant protection against ischemic brain damage despite high level poly-ADP-ribosylation. Neurochem. Res. 2017.42, 283–293
- Guofeng Lou et al. Mitophagy and Neuroprotection. Trends Mol Med. 2020 Jan;26(1):8-20.
- Yoshino, J. et al. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011.14, 528–536
- Fang EF. NAD + augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat Commun. 2019 Nov 21;10(1):5284.
- S.J. Mitchell et al. Nicotinamide improves aspects of healthspan, but not lifespan, in mice. Cell Metabol., 27 (2018), pp. 667-676
- Hector Rodriguez, Cetina Biefer, Anju Vasudevan et al. Aspects of Tryptophan and Nicotinamide Adenine Dinucleotide in Immunity: A New Twist in an Old Tale. Int J Tryptophan Res. 2017 10
- P. Bai, C. Canto, H. Oudart et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metabol., 13 (2011), pp. 461-468
- Hurtado-Bagès S, Knobloch G, Ladurner AG, Buschbeck M. The taming of PARP1 and its impact on NAD+ metabolism. Mol Metab. 2020 Aug;38:100950
- Yahyah Amana et al. Therapeutic potential of boosting NAD+ in aging and age-related diseases. Trans Med Ag. Volume 2, January 2018, Pages 30-37
- D. Kraus, Q. Yang, D. Kong, et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature, 508 (2014), pp. 258-262
- Uddin, G.M. et al. Head to head comparison of short- term treatment with the NAD(+) precursor nicotinamide mono- nucleotide (NMN) and 6 weeks of exercise in obese female mice. Front. Pharmacol. 2016.7, 258
- Mitchell, S.J. et al. Effects of sex, strain, and energy intake on hallmarks of aging in mice. Cell Metab. 2016. 23, 1093–1112
- Evandro F. Fang NAD+ in Aging: Molecular Mechanisms and Translational Implications. TRMOME Volume 23, Issue 10, October 2017, Pages 899-916
- B. Poljsak, I. Milisav. NAD+ as the link between oxidative stress, inflammation, caloric restriction, exercise, DNA repair, longevity, and health span. Rejuvenation Res., 2016. 19 (5), pp. 406-413
- S.A. Trammell, M.S. Schmidt, B.J. Weidemann, P. Redpath, F. Jaksch, R.W. Dellinger, et al. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun., 7 2016, p. 12948
- R.W. Dellinger, S.R. Santos, M. Morris, M. Evans, D. Alminana, L. Guarente, et al. Repeat dose NRPT (nicotinamide riboside and pterostilbene) increases NAD(+) levels in humans safely and sustainably: a randomized, double-blind, placebo-controlled study NPJ Aging Mech. Dis. 2017, p. 17
- C.R. Martens, B.A. Denman, M.R. Mazzo, M.L. Armstrong, N. Reisdorph, M.B. McQueen, et al. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD(+) in healthy middle-aged and older adults. Nat. Commun., 2018, p. 1286
- H.Q. Ju, Z.N. Zhuang, H. Li, T. Tian, Y.X. Lu, X.Q. Fan, et al. Regulation of the Nampt-mediated NAD salvage pathway and its therapeutic implications in pancreatic cancer. Cancer Lett. 2016, pp. 1-11




{kind=link}